





علاوه بر فناوری، سنتز گلیکوزیدها همواره مورد توجه علم بوده است، زیرا واکنشی بسیار رایج در طبیعت است. مقالات اخیر اشمیت و توشیما و تاتسوتا، و همچنین بسیاری از مراجع ذکر شده در آنها، در مورد طیف گستردهای از پتانسیلهای سنتزی اظهار نظر کردهاند.
در سنتز گلیکوزیدها، یک جزء چند قندی با یک هستهدوست مانند الکلها، کربوهیدراتها یا پروتئینها ترکیب میشود. اگر یک واکنش انتخابی با یکی از گروههای هیدروکسیل یک کربوهیدرات مورد نیاز باشد، باید تمام عملکردهای دیگر در مرحله اول محافظت شوند. در اصل، فرآیندهای آنزیمی یا میکروبی، به دلیل گزینشپذیریشان، میتوانند جایگزین مراحل پیچیده محافظت شیمیایی و محافظتزدایی شوند تا به طور انتخابی از گلیکوزیدها در مناطق مختلف جدا شوند. با این حال، به دلیل سابقه طولانی آلکیل گلیکوزیدها، کاربرد آنزیمها در سنتز گلیکوزیدها به طور گسترده مورد مطالعه و کاربرد قرار نگرفته است.
با توجه به ظرفیت سیستمهای آنزیمی مناسب و هزینههای بالای تولید، سنتز آنزیمی آلکیل پلیگلیکوزیدها برای ارتقاء به سطح صنعتی آماده نیست و روشهای شیمیایی ترجیح داده میشوند.
در سال 1870، مککالی سنتز «استوکلروهیدروز» (1، شکل 2) را از طریق واکنش دکستروز (گلوکز) با استیل کلرید گزارش کرد، که در نهایت منجر به تاریخچه مسیرهای سنتز گلیکوزیدها شد.
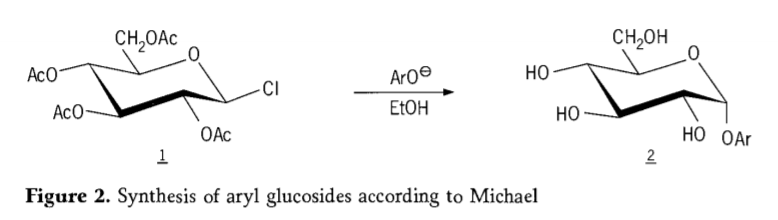
بعدها مشخص شد که تترا-0-استیل-گلوکوپیرانوزیل هالیدها (استوهالوگلوکزها) واسطههای مفیدی برای سنتز استریوگزین آلکیل گلوکوزیدهای خالص هستند. در سال 1879، آرتور مایکل موفق به تهیه آریل گلیکوزیدهای مشخص و قابل تبلور از واسطههای کولی و فنولاتها شد. (شکل 2).
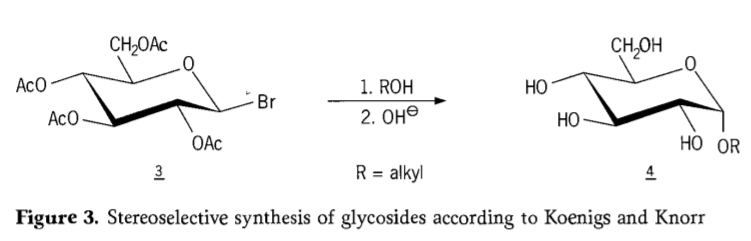
در سال ۱۹۰۱، سنتز مایکل برای طیف وسیعی از کربوهیدراتها و آگلیکونهای هیدروکسیلی، زمانی که W.Koenigs و E.Knorr فرآیند بهبود یافتهی گلیکوزیداسیون استریوسلکتیو خود را معرفی کردند (شکل ۳)، انجام شد. این واکنش شامل جایگزینی SN2 در کربن آنومری است و به صورت استریوسلکتیو با وارونگی پیکربندی پیش میرود و به عنوان مثال، α-گلوکوزید ۴ را از β-آنومر واسطهی آسئوبرموگلوکز ۳ تولید میکند. سنتز کونیگز-کنور در حضور پیشبرندههای نقره یا جیوه انجام میشود.
در سال ۱۸۹۳، امیل فیشر رویکردی اساساً متفاوت برای سنتز آلکیل گلوکوزیدها پیشنهاد کرد. این فرآیند اکنون به عنوان "گلیکوزیداسیون فیشر" شناخته میشود و شامل واکنش کاتالیز شده اسیدی گلیکوزها با الکلها است. با این وجود، هر گونه گزارش تاریخی باید شامل اولین تلاش گزارش شده A.Gautier در سال ۱۸۷۴ برای تبدیل دکستروز با اتانول بیآب در حضور اسید هیدروکلریک نیز باشد. به دلیل یک تجزیه و تحلیل عنصری گمراهکننده، گوتیه معتقد بود که "دیگلوکز" به دست آورده است. فیشر بعداً نشان داد که "دیگلوکز" گوتیه در واقع عمدتاً اتیل گلوکوزید است (شکل ۴).
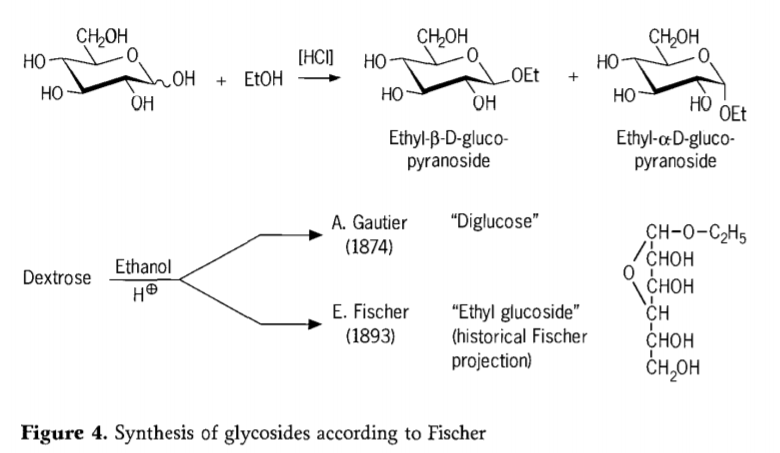
فیشر ساختار اتیل گلوکوزید را به درستی تعریف کرد، همانطور که از فرمول تاریخی فورانوزیدی پیشنهادی میتوان دریافت. در واقع، محصولات گلیکوزیداسیون فیشر پیچیده هستند، عمدتاً مخلوطهای تعادلی از α/β-آنومرها و ایزومرهای پیرانوزید/فورانوزید که شامل الیگومرهای گلیکوزیدی با پیوند تصادفی نیز میباشند.
بر این اساس، جداسازی گونههای مولکولی منفرد از مخلوط واکنش فیشر آسان نیست، که در گذشته یک مشکل جدی بوده است. پس از بهبود این روش سنتز، فیشر متعاقباً سنتز کونیگز-کنور را برای تحقیقات خود اتخاذ کرد. با استفاده از این فرآیند، ای. فیشر و بی. هلفریچ اولین کسانی بودند که در سال ۱۹۱۱ سنتز یک آلکیل گلوکوزید با زنجیره بلند را که خواص سورفکتانت از خود نشان میداد، گزارش کردند.
فیشر در اوایل سال ۱۸۹۳ به درستی متوجه خواص اساسی آلکیل گلیکوزیدها، مانند پایداری بالای آنها در برابر اکسیداسیون و هیدرولیز، به ویژه در محیطهای قلیایی قوی، شده بود. هر دو ویژگی برای پلیگلیکوزیدهای آلکیل در کاربردهای سورفکتانت ارزشمند هستند.
تحقیقات مربوط به واکنش گلیکوزیداسیون هنوز در حال انجام است و چندین مسیر جالب برای تولید گلیکوزیدها در گذشته اخیر توسعه یافته است. برخی از روشهای سنتز گلیکوزیدها در شکل 5 خلاصه شدهاند.
به طور کلی، فرآیندهای گلیکوزیداسیون شیمیایی را میتوان به فرآیندهایی تقسیم کرد که منجر به تعادل پیچیده الیگومر در تبادل گلیکوزیل کاتالیز شده با اسید میشوند.
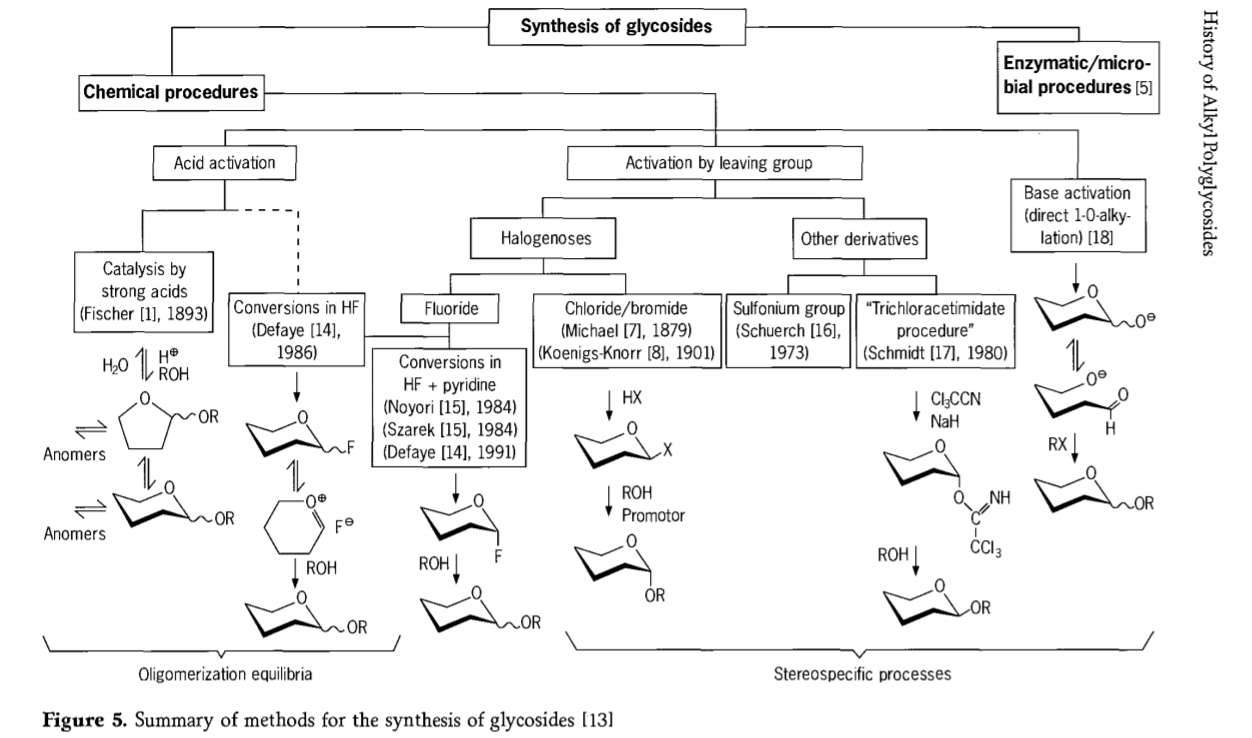
واکنشها روی سوبستراهای کربوهیدراتی که به طور مناسب فعال شدهاند (واکنشهای گلیکوزیدی فیشر و واکنشهای هیدروژن فلوراید (HF) با مولکولهای کربوهیدرات محافظت نشده) و واکنشهای جانشینی کنترلشده، برگشتناپذیر و عمدتاً استریوتاکسی با سینتیک. نوع دوم روش ممکن است منجر به تشکیل گونههای منفرد به جای مخلوطهای پیچیده واکنشها شود، به ویژه هنگامی که با تکنیکهای گروههای پایستگی ترکیب شود. کربوهیدراتها ممکن است گروههایی را روی کربن نابجا، مانند اتمهای هالوژن، سولفونیلها یا گروههای تریکلرواستیمیدات، باقی بگذارند یا قبل از تبدیل به استرهای تریفلات توسط بازها فعال شوند.
در مورد خاص گلیکوزیداسیونها در هیدروژن فلوراید یا در مخلوطهای هیدروژن فلوراید و پیریدین (پیریدینیوم پلی [هیدروژن فلوراید])، گلیکوزیل فلورایدها درجا تشکیل میشوند و به آرامی به گلیکوزیدها تبدیل میشوند، به عنوان مثال با الکلها. نشان داده شده است که هیدروژن فلوراید یک محیط واکنش قویاً فعالکننده و غیرتجزیهکننده است. تراکم خودکار تعادلی (الیگومریزاسیون) مشابه فرآیند فیشر مشاهده میشود، اگرچه مکانیسم واکنش احتمالاً متفاوت است.
آلکیل گلیکوزیدهای خالص شیمیایی فقط برای کاربردهای بسیار خاص مناسب هستند. به عنوان مثال، آلکیل گلیکوزیدها با موفقیت در تحقیقات بیوشیمیایی برای تبلور پروتئینهای غشایی، مانند تبلور سهبعدی پورین و باکتریورودوپسین در حضور اکتیل β-D-گلوکوپیرانوزید، مورد استفاده قرار گرفتهاند (آزمایشهای بیشتر بر اساس این کار منجر به جایزه نوبل شیمی برای دایزنهوفر، هوبر و میشل در سال ۱۹۸۸ شد).
در طول توسعه پلیگلیکوزیدهای آلکیل، روشهای استریوگزین در مقیاس آزمایشگاهی برای سنتز انواع مواد مدل و مطالعه خواص فیزیکوشیمیایی آنها مورد استفاده قرار گرفتهاند، اما به دلیل پیچیدگی آنها، ناپایداری واسطهها و مقدار و ماهیت بحرانی ضایعات فرآیند، سنتزهای نوع کونیگز-کنور و سایر تکنیکهای گروه محافظ، مشکلات فنی و اقتصادی قابل توجهی ایجاد میکنند. فرآیندهای نوع فیشر نسبتاً کمتر پیچیده و در مقیاس تجاری آسانتر انجام میشوند و بر این اساس، روش ترجیحی برای تولید پلیگلیکوزیدهای آلکیل در مقیاس بزرگ هستند.
زمان ارسال: ۱۲ سپتامبر ۲۰۲۰